physiologie normale de l’aldostérone
L’aldostérone participe à l’homéostasie du volume sanguin circulant et de la concentration sérique en potassium; ceux-ci, à leur tour, se nourrissent pour réguler la sécrétion d’aldostérone par la zone glomérulaire du cortex surrénalien. La sécrétion d’aldostérone est stimulée par une déplétion réelle ou apparente du volume sanguin détectée par les récepteurs d’étirement et par une augmentation des concentrations sériques d’ions potassium; elle est supprimée par l’hypervolémie et l’hypokaliémie.,
Les mécanismes de régulation de la sécrétion d’aldostérone sont complexes, impliquant la zone glomérulaire des glandes surrénales, l’appareil juxtaglomérulaire des reins, le système cardiovasculaire, le système nerveux autonome, les poumons et le foie (voir l’image ci-dessous). Les principaux facteurs qui stimulent la production et la libération d’aldostérone par la zona glomérulosa sont l’angiotensine II et la concentration sérique de potassium. L’appareil juxtaglomérulaire est le principal site de régulation de la production d’angiotensine II.,
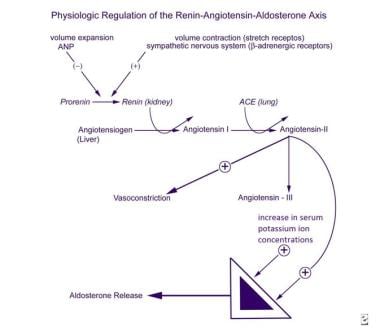 régulation Physiologique du système rénine-angiotensine-aldostérone.
régulation Physiologique du système rénine-angiotensine-aldostérone. L’ACTH stimule la sécrétion d’aldostérone de manière aiguë et transitoire, mais ne semble pas jouer un rôle significatif dans la régulation à long terme de la sécrétion de minéralocorticoïdes. Les principaux inhibiteurs de la zona glomérulosa comprennent le peptide natriurétique auriculaire circulant (ANP) et, localement, la dopamine., Bien que les niveaux D’ANP soient clairement augmentés dans l’hyperaldostéronisme, ni L’ANP ni la dopamine n’ont été impliqués comme cause principale de la sécrétion d’aldostérone cliniquement désordonnée.
le métoclopramide a été montré pour augmenter la sécrétion d’aldostérone, suggérant que la dopamine peut inhiber toniquement la libération d’aldostérone. Les rôles physiologiques de l’adrénomédulline et du peptide intestinal vasoactif (VIP) sur la sécrétion d’aldostérone restent à clarifier, bien que ces deux neuropeptides soient produits chez le rat zona glomérulosa.,
la synthèse de la prorénine, sa conversion en rénine et sa sécrétion systémique sont stimulées par la contraction du volume sanguin détectée par les récepteurs d’étirement, la stimulation bêta-adrénergique du système nerveux sympathique et les prostaglandines I2 et E2. Ces processus sont inhibés par l’expansion du volume et ANP.
la rénine convertit l’angiotensinogène, un proenzyme synthétisé dans le foie, en le décapeptide angiotensine I, qui est ensuite converti dans les poumons en l’octapeptide angiotensine II par l’enzyme de conversion de l’angiotensine (ACE)., L’angiotensine II est à la fois un stimulateur de la sécrétion d’aldostérone et un puissant vasopresseur. L’angiotensine II est métabolisée en angiotensine III, un heptapeptide qui est également un stimulateur de la sécrétion d’aldostérone.
la synthèse et la sécrétion des prostaglandines I2 et E2 et la fonction normale des récepteurs d’étirement dépendent de la concentration de calcium ionisé intracellulaire. La sécrétion rénale de prostaglandine est stimulée par les catécholamines et l’angiotensine II., La régulation complexe de la synthèse et de la sécrétion d’aldostérone fournit plusieurs points auxquels une perturbation de la régulation de la sécrétion d’aldostérone peut se produire.
L’aldostérone est synthétisée à partir du cholestérol en une série de 6 étapes biosynthétiques (voir l’image ci-dessous). Seules les 2 dernières étapes sont spécifiques à la synthèse de l’aldostérone; les 4 premières s’appliquent également à la synthèse du cortisol par la zona fasciculata. Par conséquent, un défaut dans l’une des enzymes synthétiques spécifiques de l’aldostérone ne conduit pas à l’hypercortisolisme et à l’hyperplasie surrénale secondaire médiée par L’ACTH.,
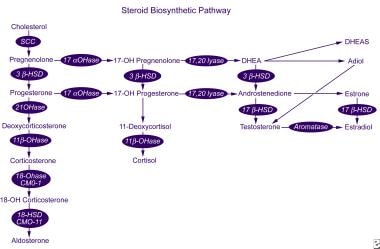 voie de biosynthèse des Stéroïdes.
voie de biosynthèse des Stéroïdes. l’enzyme aldostérone synthase est codée par le gène CYP11B2 et a une activité 11β-hydroxylase, 18-hydroxylase et 18-hydroxydéhydrogénase. Ce gène est situé sur le bras chromosomique humain 8q24.3-tel, à proximité du gène CYP11B1, qui code la 11β-hydroxylase, l’enzyme qui catalyse l’étape finale de la synthèse du cortisol. Des Mutations dans ces gènes peuvent entraîner un certain nombre de troubles de la synthèse de l’aldostérone (voir différentiels).,
L’action de L’aldostérone sur les tissus cibles (par exemple, le tubule rénal distal, les glandes sudoripares, les glandes salivaires et l’épithélium du gros intestin) est médiée par un récepteur minéralocorticoïde spécifique. Les récepteurs minéralocorticoïdes présentent une affinité égale pour les minéralocorticoïdes et le cortisol, mais les récepteurs de l’aldostérone dans le tubule distal et ailleurs sont protégés contre l’activation médiée par le cortisol par la 11β-hydroxystéroïde déshydrogénase de type 2, qui convertit localement le cortisol en cortisone inactive.,
aldostéronisme primaire
le terme hyperaldostéronisme primaire (ou aldostéronisme primaire ) fait référence à une augmentation indépendante de la rénine de la sécrétion d’aldostérone. Cette maladie est principalement une maladie de l’âge adulte, avec son incidence maximale dans les quatrième à sixième décennies de la vie.
plus de 90% des cas d’AP sont dus soit à un adénome produisant de l’aldostérone (APA), qui représente environ 35% des cas (30-40%), soit à un hyperaldostéronisme idiopathique (IHA), qui représente environ 60% des cas (presque tous bilatéraux)., L’hyperplasie unilatérale des surrénales (UAH) est une cause rare de PA, représentant 1 à 2% des cas. Environ 1% des patients présentent des carcinomes corticosurrénaux purement sécréteurs d’aldostérone et généralement importants au moment du diagnostic; 1% présentent un hyperaldostéronisme familial et 1% un adénome ou un carcinome ectopique produisant de l’aldostérone.
l’hyperplasie unilatérale des surrénales représente 14 à 17% de tous les cas d’AP unilatérale. La prévalence de l’adénome cortical dans l’hyperplasie corticale est estimée à 6-24%., La présentation clinique et les résultats des patients atteints d’hyperaldostéronisme primaire unilatéral sont similaires quel que soit le diagnostic histopathologique. L’hyperplasie corticosurrénale unilatérale est rare.
Les APA (parfois appelés aldostéronomes) sont généralement des adénomes bénins encapsulés de moins de 2 cm de diamètre. La plupart des cas sont solitaires, bien que dans un tiers des cas, il existe des preuves de nodularité dans la même glande surrénale, suggérant que la condition est apparue dans une glande précédemment hyperplasique.,
Les Patients atteints D’IHA présentent un épaississement bilatéral et une nodularité variable de leur cortex surrénalien. Un large spectre de gravité existe pour ce trouble, qui peut passer inaperçu pendant de longues périodes sans hypokaliémie et seulement une hypertension légère. Il a été suggéré que L’IHA résulte d’un facteur stimulant le cortex surrénalien non détecté. Alternativement, le trouble peut survenir à la suite d’une mutation activante dans un gène spécifique du cortex surrénalien. Aucune des deux hypothèses n’a été prouvée.,
Les formes héréditaires d’hyperaldostéronisme primaire ne représentent que 1% des cas, mais sont plus susceptibles de survenir pendant l’enfance. Ces formes comprennent l’hyperaldostéronisme familial (FH) de types I, II et III.
l’hyperaldostéronisme Familial de type I
L’hyperaldostéronisme familial de type I (FH-I), également appelé aldostéronisme glucocorticoïde réparable (GRA), peut être détecté chez des individus asymptomatiques lors du dépistage de la progéniture des individus affectés, ou les patients peuvent présenter dans pour prospérer en raison de l’hypokaliémie., La FH-I est héritée de manière autosomique dominante et présente une faible fréquence de nouvelles mutations.
la première description clinique de la GRA est apparue en 1966 et le mécanisme génétique a été découvert en 1992. La FH-I résulte d’un croisement inégal de la CYP11B1 (gène de la 11β-hydroxylase) et de la CYP11B2 (gène de l’aldostérone synthase) hautement apparentés au cours de la méiose, produisant un produit de fusion de type anti-Lépore., Ce réarrangement génétique fait que l’expression du CYP11B2 est placée sous le contrôle du promoteur du CYP11B1 et que la synthèse de l’aldostérone est anormalement régulée par L’ACTH plutôt que par le système rénine-angiotensine.
le résultat est la production D’aldostérone dépendante de L’ACTH et la production d’analogues 17-hydroxylés du 18-hydroxycortisol sous régulation de L’ACTH à partir de l’expression enzymatique ectopique dans la zona fasciculata. Une hyperplasie bilatérale de la zona fasciculata se produit et des niveaux élevés de nouveaux 18-hydroxystéroïdes apparaissent dans l’urine., La formation d’adénomes est rare, mais les patients ont une augmentation significative de l’incidence des anévrismes cérébrovasculaires, pour lesquels ils nécessitent un dépistage.
hyperaldostéronisme Familial de type II
FH type II (FH-II) est une forme héréditaire Non glucocorticoïde suppressible de l’hyperaldostéronisme qui a d’abord été reconnu comme une entité distincte par Gordon et al, bien que des cas aient déjà été décrits dans les années 1980. comme FH–I, il est hérité de manière autosomique dominante. Contrairement à FH-I, certains types de FH-II présentent un taux élevé de formation d’adénomes.,
le mécanisme et le locus génique n’ont pas encore été identifiés, bien que le CYP11B et les gènes récepteurs de la rénine et de l’angiotensine II aient été exclus. Cependant, un couplage a été établi pour un certain nombre de familles à la bande 7p22. Il a également été spéculé que FH-II n’est pas un trouble unique.
hyperaldostéronisme Familial de type III
le FH-III est une forme autosomique dominante rare de PA caractérisée par une hypertension précoce, un hyperaldostéronisme Non-glucocorticoïde réparable et une hypokaliémie., Des mutations hétérozygotes de la lignée germinale du gène KCNJ5, codant Kir3.4, un membre de la famille des canaux K+ rectifiant vers l’intérieur, ont été identifiées comme une cause de FH-III. Jusqu’à présent, 4 mutations (G151R, G151E, T158A et I157S) ont été rapportées dans 6 familles.
Le phénotype clinique des patients porteurs des mutations ci-dessus varie de L’AP sévère et de l’hypertension réfractaire au traitement médical nécessitant une surrénalectomie bilatérale, à l’hypertension légère ou modérée sensible au traitement médical. Chez certains patients, une hyperplasie surrénale a été décrite.,
diverses études de différents centres rapportent une prévalence de mutations somatiques KCNJ5 dans les apa sporadiques allant de 30 à 65%. Il y a 2 mutations récurrentes, G151R et L168R, rapportées par toutes les études, alors qu’il y a un rapport d’une délétion de 3 nucléotides, le delI157.
Les résidus affectés de la lignée germinale et des mutations somatiques se trouvent dans ou à proximité du filtre de sélectivité du canal potassique Kir3.4 et sont hautement conservés chez différentes espèces., Des études électrophysiologiques démontrent que ces mutations entraînent une perte de sélectivité des canaux, avec une augmentation de la conductance Na+ conduisant à une dépolarisation de la membrane. Dans les cellules de la zona glomérulosa, la dépolarisation membranaire conduit à l’ouverture de canaux Ca2+ activés par tension, avec activation de la voie de signalisation calcique, le principal médiateur de la production d’aldostérone.
Les APA avec mutations KCNJ5 sont plus répandus chez les femmes que chez les hommes et chez les patients plus jeunes. Ils sont également associés à des niveaux d’aldostérone préopératoires plus élevés., Ils ne sont pas liés à la taille de la tumeur, mais ils sont liés à des niveaux d’aldostérone plus élevés et à des concentrations de K+ plus faibles.
Les analyses du Transcriptome et de la réaction en chaîne par polymérase (PCR) en temps réel démontrent que les apa avec des mutations KCNJ5 présentent une expression accrue du gène CYP11B2 et de son régulateur transcriptionnel NR4A2, augmentant ainsi la production d’aldostérone. Il a également été constaté que les apa avec et sans mutations KCNJ5 présentent des modèles d’expression génique légèrement différents., Une autre étude rapporte des niveaux D’ARNm KCNJ5 plus élevés dans les apa avec des mutations KCNJ5 et significativement plus élevés dans les apa que les adénomes et les phéochromocytomes producteurs de cortisol.
mutations somatiques de L’ATP1A1 (gène codant pour la sous-unité alpha-1 de L’ATPase Na+ / K+, un membre de la famille des ATPases de type P), de L’ATP2B3 (gène codant pour le calcium de la membrane plasmique transportant L’ATPase 3 , un autre membre de la famille des ATPases de,3, la sous-unité alpha d’un canal calcique voltage-gated de type L) sont présents dans environ 6%, 1% et 8% de tous les cas d’un adénome produisant de l’aldostérone, respectivement. Plus récemment, des mutations de novo dans la lignée germinale de CACNA1D ont été rapportées chez 2 enfants atteints d’un syndrome non décrit qui présentait des anomalies PA et neuromusculaires.,
hyperaldostéronisme secondaire
l’hyperaldostéronisme secondaire est un terme collectif pour un groupe diversifié de troubles caractérisés par l’activation physiologique de l’axe rénine-angiotensine-aldostérone (R-A-A) en tant que mécanisme homéostatique conçu pour maintenir les concentrations d’électrolytes sériques ou le volume de liquide. En présence d’une fonction rénale normale, elle peut entraîner une hypokaliémie.
l’hyperaldostéronisme secondaire peut être divisé en 2 catégories, 1 avec hypertension associée et 1 sans., La première catégorie comprend l’hypertension rénovasculaire, qui résulte d’une ischémie rénale et d’une hypoperfusion conduisant à l’activation de l’axe R-A-A. Les causes les plus fréquentes de sténose de l’artère rénale chez les enfants sont l’hyperplasie fibromusculaire et la neurofibromatose. Une hypokaliémie peut survenir chez 20% des patients.
Les taux D’activité rénine plasmatique (ARP) se situent souvent dans la plage de référence, mais des taux élevés d’ARP peuvent être détectés après provocation avec une dose unique de captopril 1 mg/kg., On pense également que l’ischémie rénale sous-tend l’hyperaldostéronisme secondaire observé dans l’hypertension maligne.
Une Hyperréninémie et un aldostéronisme secondaire ont également été rapportés chez des patients atteints de phéochromocytome, apparemment à la suite d’une sténose fonctionnelle de l’artère rénale. Les tumeurs productrices de rénine sont très rares et des niveaux très élevés de PRA (jusqu’à 50 ng/mL/h) sont notés, fréquemment avec un rapport prorénine / rénine accru. Les tumeurs sont généralement d’origine rénale et comprennent les tumeurs de Wilms et carcinomes rénaux.,
l’Hyperkaliémie due à une insuffisance rénale chronique provoque aussi un hyperaldostéronisme secondaire. De faibles ratios sodium / potassium peuvent être mesurés dans la salive et les selles. L’hypertension induite par la Cyclosporine chez les patients transplantés d’organes solides peut également impliquer un composant de l’hyperaldostéronisme.
l’hyperaldostéronisme secondaire en l’absence d’hypertension survient à la suite de tentatives homéostatiques de maintenir la concentration de sodium ou le volume circulatoire ou de réduire la concentration de potassium., Les conditions cliniques dans lesquelles il peut survenir comprennent la diarrhée, la transpiration excessive, un faible débit cardiaque et une hypoalbuminémie due à une maladie hépatique ou rénale ou à un syndrome néphrotique. L’hyperaldostéronisme secondaire peut également survenir au niveau du développement chez les nouveau-nés (voir ci-dessous).
augmentation de la dépendance aux minéralocorticoïdes chez les jeunes
la dépendance aux minéralocorticoïdes de la réabsorption du sodium augmente pendant la petite enfance et l’enfance, atteignant un sommet pendant la période néonatale avant de diminuer progressivement avec l’âge., Cette augmentation se produit parce que la réabsorption du sodium et de l’eau par le tubule proximal est moins efficace au début de la vie, ce qui entraîne une augmentation de la charge en sodium et en eau au niveau du tubule rénal distal.
étant donné que la résorption du sodium et de l’eau du tubule distal est médiée par L’axe R-A-A, l’ARP est environ 10 à 20 fois plus élevée chez un nouveau-né que chez un ADULTE., Par conséquent, les nouveau-nés montrent des augmentations relatives des taux de production d’aldostérone (>300 µg/m2/jour vs 50 µg/m2/jour chez un adulte) et des concentrations plasmatiques d’aldostérone (80 PG/DL vs 16 pg/dL). Ces augmentations au début de la vie expliquent pourquoi les jeunes nourrissons présentent de profonds symptômes cliniques d’hyperaldostéronisme qui s’améliorent progressivement avec l’âge.