Normal aldosterona fisiologia
Aldosterona participa da homeostase do volume de sangue circulante e a concentração de potássio sérico; estes, por sua vez, feed back para regular a secreção de aldosterona pela zona glomerulosa do córtex adrenal. A secreção de aldosterona é estimulada por uma depleção real ou aparente no volume sanguíneo detectada pelos receptores esticadores e por um aumento nas concentrações séricas de iões de potássio; é suprimida pela hipervolemia e hipocaliemia.,
Os mecanismos que regulam a secreção de aldosterona são complexos, envolvendo a zona glomerulosa das glândulas supra-renais, o juxtaglomerular aparelho nos rins, sistema cardiovascular, sistema nervoso autônomo, os pulmões e o fígado (ver imagem abaixo). Os principais factores que estimulam a produção e libertação de aldosterona pela zona glomerulosa são a angiotensina II e a concentração sérica de potássio. O aparelho justaglomerular é o principal local de regulação da produção de angiotensina II.,
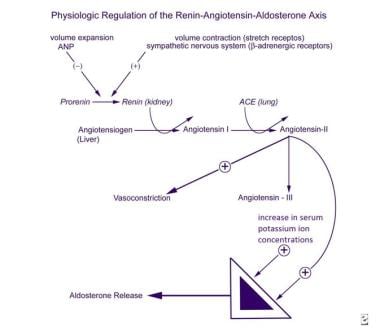 regulação fisiológica do eixo renina-angiotensina-aldosterona.
regulação fisiológica do eixo renina-angiotensina-aldosterona. ACTH estimula a secreção de aldosterona de forma aguda e transitória, mas não parece desempenhar um papel significativo na regulação a longo prazo da secreção mineralocorticóide. Os principais inibidores da zona glomerulosa incluem o peptídeo natriurético Auricular circulante (ANP) e, localmente, a dopamina., Embora os níveis de ANP estejam claramente aumentados no hiperaldosteronismo, nem a ANP nem a dopamina foram implicadas como causa primária da secreção de aldosterona clinicamente desordenada.
metoclopramida demonstrou aumentar a secreção de aldosterona, sugerindo que a dopamina pode inibir tonicamente a libertação de aldosterona. Os papéis fisiológicos da adrenomedullin e do peptídeo intestinal vasoactivo (VIP) na secreção de aldosterona continuam por esclarecer, embora ambos os neuropeptídeos sejam produzidos na zona glomerulosa do rato.,
a síntese da prorenina, a sua conversão à renina, e a sua secreção sistémica são estimulados pela contracção do volume sanguíneo detectada pelos receptores esticadores, pela estimulação beta-adrenérgica do sistema nervoso simpático e pelas prostaglandinas I2 e E2. Estes processos são inibidos pela expansão de volume e ANP.
renina converte o angiotensinogénio, uma proenzima sintetizada no fígado, no decapéptido angiotensina I, que é então convertido nos pulmões no octapeptídeo angiotensina II pela enzima de conversão da angiotensina (ECA)., A angiotensina II é simultaneamente um estimulador da secreção de aldosterona e um vasopressor potente. A angiotensina II é metabolizada em angiotensina III, um heptapéptido que é também um estimulador da secreção de aldosterona.
a síntese e secreção das prostaglandinas I2 e E2 e a função normal dos receptores de estiramento são dependentes da concentração intracelular de cálcio ionizado. A secreção Renal de prostaglandinas é estimulada pelas catecolaminas e pela angiotensina II., A regulação complexa da síntese e secreção da aldosterona fornece vários pontos nos quais pode ocorrer perturbação na regulação da secreção de aldosterona.a aldosterona é sintetizada a partir do colesterol numa série de 6 passos biossintéticos (ver a imagem abaixo). Apenas os últimos 2 passos são específicos à síntese da aldosterona; os primeiros 4 também se aplicam à síntese do cortisol pela zona fasciculata. Consequentemente, um defeito numa das enzimas sintéticas específicas da aldosterona não conduz a hipercortisolismo e hiperplasia adrenal secundária mediada pela ACTH.,
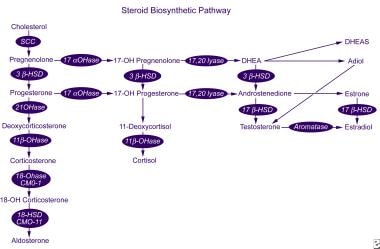 via biossintética dos esteróides.
via biossintética dos esteróides. A enzima aldosterona sintase é codificado pelo gene CYP11B2 e tem 11β-hydroxylase, 18-hydroxylase, e 18-hydroxydehydrogenase atividade. Este gene está localizado no braço do cromossomo humano 8q24.3-tel, perto do gene CYP11B1, que codifica 11β-hidroxilase, a enzima que catalisa o passo final da síntese do cortisol. Mutações nestes genes podem resultar em uma série de distúrbios da síntese da aldosterona (ver diferenciais)., a acção da aldosterona nos tecidos alvo (por exemplo, o túbulo renal distal, glândulas sudoríparas, glândulas salivares e epitélio do intestino grosso) é mediada através de um receptor mineralocorticóide específico. Os receptores mineralocorticóides apresentam afinidade igual para os mineralocorticóides e o cortisol, mas os receptores da aldosterona no túbulo distal e noutros locais estão protegidos da activação mediada pelo cortisol pela desidrogenase tipo 2 do 11β-hidroxisteróide, que localmente converte o cortisol em cortisona inactiva.,aldosteronismo primário o termo hiperaldosteronismo primário (ou aldosteronismo primário ) refere-se a um aumento independente da renina na secreção da aldosterona. Esta condição é principalmente uma doença da idade adulta, com sua incidência máxima nas quarta a sexta décadas de vida.
Mais de 90% dos casos de PA são devido a uma aldosterona-produção de adenoma (APA), que representa cerca de 35% dos casos (30-40%), ou idiopática hiperaldosteronismo (IHA), que representa cerca de 60% dos casos (quase todos são bilaterais)., Hiperplasia adrenal Unilateral (UAH) é uma causa rara de PA, sendo responsável por 1-2% dos casos. Cerca de 1% dos pacientes apresentam carcinomas adrenocorticais que são puramente secreções da aldosterona e são geralmente grandes no momento do diagnóstico; 1% apresentam hiperaldosteronismo familiar, e 1% apresentam um adenoma ou carcinoma que produz aldosterona ectópica. a hiperplasia supra-renal Unilateral representa 14-17% de todos os casos de PA unilateral. Estima-se que a prevalência de adenoma cortical dentro da hiperplasia cortical seja de 6-24%., A apresentação clínica e o resultado dos doentes com hiperaldosteronismo primário unilateral são semelhantes independentemente do diagnóstico histopatológico. Hiperplasia adrenocortical Unilateral é rara.
APAs (por vezes referido como aldosteronomas) são geralmente adenomas encapsulados benignos com menos de 2 cm de diâmetro. A maioria dos casos são solitários, embora em um terço dos casos, existam evidências de nodularidade na mesma glândula adrenal, sugerindo que a condição surgiu em uma glândula hiperplástica anteriormente.,
doentes com IHA têm espessamento bilateral e nodularidade variável do córtex supra-renal. Existe um amplo espectro de gravidade para esta doença, que pode ir indetectável por longos períodos sem hipocaliemia e apenas hipertensão ligeira. Tem sido sugerido que a IHA surge como resultado de um fator não detectado de estimulação do córtex adrenal. Alternativamente, a doença pode surgir como resultado de uma mutação activadora num gene específico do córtex supra–renal. Nenhuma hipótese foi provada.,
formas hereditárias de hiperaldosteronismo primário representam apenas 1% dos casos, mas são mais prováveis de ocorrer durante os anos de infância. Estas formas incluem familiar hiperaldosteronismo (FH) tipos I, II e III.
Familiares hiperaldosteronismo tipo I
FH tipo I (FH-I), também conhecido como glicocorticóides-remediable aldosteronism (GRA), podem ser detectados em indivíduos assintomáticos durante o rastreio da prole de indivíduos afetados, ou pacientes podem se apresentar na infância com hipertensão, fraqueza, e dificuldade de crescimento, devido à hipocalemia., O FH-I é herdado de forma autossómica dominante e tem uma baixa frequência de novas mutações.
a primeira descrição clínica do GRA apareceu em 1966, e o mecanismo genético foi descoberto em 1992. O FH-I surge como resultado da passagem desigual de CYP11B1 (o gene 11β-hidroxilase) e CYP11B2 (o gene da aldosterona sintase) durante a meiose, produzindo um produto de fusão do tipo Anti-leporo., Este rearranjo genético faz com que a expressão do CYP11B2 seja colocada sob o controlo do promotor CYP11B1 e a síntese da aldosterona seja anormalmente regulada pelo ACTH e não pelo sistema renina-angiotensina.
o resultado é a produção de aldosterona dependente de ACTH e a produção de análogos 17-hidroxilados de 18-hidroxicortisol ao abrigo do regulamento ACTH a partir da expressão enzimática ectópica na zona fasciculata. Ocorre hiperplasia Bilateral da zona fasciculata, e altos níveis de novos 18-hidroxisteróides aparecem na urina., A formação de Adenoma é rara, mas os doentes têm um aumento significativo na incidência de aneurismas cerebrovasculares, para os quais necessitam de rastreio.
Familiares hiperaldosteronismo tipo II
FH tipo II (FH-II) não é um glicocorticóide suppressible formulário herdado do hiperaldosteronismo que foi reconhecido pela primeira vez como uma entidade distinta, por Gordon et al, embora casos haviam sido previamente descritas na década de 1980. Como FH-eu, é herdada de forma autossômica dominante maneira. Em contraste com FH-I, algumas famílias FH-II exibem uma alta taxa de formação de adenoma.,
o mecanismo e o gene locus ainda não foram identificados, embora tenham sido excluídos os genes do CYP11B e do receptor da renina e da angiotensina II. No entanto, a ligação foi estabelecida para uma série de famílias para a banda 7p22. Também foi especulado que o FH-II não é um único transtorno.
Familiares hiperaldosteronismo tipo III
FH-III é uma rara autossômica dominante formulário do PA, caracterizada por início precoce da hipertensão, nonglucocorticoid-remediable hiperaldosteronismo, e hipocalemia., Germinativa heterozigotos mutações com perda de sentido do KCNJ5 gene, que codifica Kir3.4, um membro do interiormente rectificação K+ canal de família, tem sido identificado como uma causa de FH-III. Até agora, 4 mutações (G151R, G151E, T158A, e I157S) têm sido relatados em 6 famílias.
o fenótipo clínico dos doentes que abrigam as mutações acima varia desde a PA grave e a hipertensão refractária ao tratamento médico que requer adrenalectomia bilateral, à hipertensão ligeira ou moderada que responde à terapêutica médica. Em alguns doentes foi descrita hiperplasia supra-renal.,
vários estudos de diferentes centros relatam uma prevalência de mutações somáticas do KCNJ5 em APAs esporádicas variando de 30-65%. Existem 2 mutações recorrentes, G151R e L168R, notificadas por todos os estudos, ao passo que existe um relatório de eliminação de 3 nucleótidos, a delI157.
os resíduos afectados tanto da germina como das mutações somáticas estão dentro ou perto do filtro de selectividade do canal de potássio Kir3.4 e são altamente conservados entre diferentes espécies., Estudos electrofisiológicos demonstram que estas mutações resultam na perda de selectividade do canal, com o aumento da condutância na+ levando à despolarização da membrana. Nas células da zona glomerulosa, a despolarização das membranas leva à abertura de canais Ca2+ activados por voltagem, com a activação da via de sinalização do cálcio, o principal mediador da produção de aldosterona.APAs com mutações KCNJ5 são mais prevalentes nas mulheres do que nos homens e em doentes mais jovens. Estão também associados a níveis pré-operatórios mais elevados de aldosterona., Não estão relacionados com o tamanho do tumor, mas estão relacionados com níveis mais elevados de aldosterona e concentrações mais baixas de K+.as análises de transcriptoma e de reacção em cadeia da polimerase em tempo real (PCR) demonstram que as mutações APAs com mutações KCNJ5 exibem uma expressão aumentada do gene CYP11B2 e do seu regulador transcritional NR4A2, aumentando assim a produção de aldosterona. Também foi descoberto que o APAs com e sem mutações KCNJ5 exibem padrões de expressão genética ligeiramente diferentes., Outro estudo relata níveis de mRNA KCNJ5 mais elevados nos APAs com mutações KCNJ5 e significativamente mais elevados nos APA do que os adenomas produtores de cortisol e os feocromocitomas.
mutações Somáticas no ATP1A1 (gene que codifica a alfa-1 subunidade da Na+/K+ ATPase, um membro do tipo P ATPase família), ATP2B3 (gene que codifica a membrana plasmática de cálcio transporte ATPase 3 , outro membro do tipo P ATPase da família), ou CACNA1D (gene que codifica Cav1.,3, a subunidade alfa de um canal de Cálcio Revestido por tensão do tipo L) está presente em aproximadamente 6%, 1% e 8% de todos os casos de adenoma produtor de aldosterona, respectivamente. Mais recentemente, foram notificadas mutações de novo germline em CACNA1D em 2 crianças com uma síndrome não descrita anteriormente que apresentava anomalias PA e neuromuscular., hiperaldosteronismo secundário hiperaldosteronismo secundário é um termo colectivo para um grupo diverso de perturbações caracterizadas pela activação fisiológica do eixo renina-angiotensina-aldosterona (R-A-A) como mecanismo homeostático concebido para manter as concentrações séricas de electrólitos ou o volume de fluido. Na presença de função renal normal, pode levar a hipocaliemia.
hiperaldosteronismo secundário pode ser dividido em 2 categorias, 1 com hipertensão associada e 1 sem., A primeira categoria inclui a hipertensão renovascular, que resulta de isquemia renal e hipoperfusão levando à ativação do eixo R-A-A. As causas mais comuns de estenose da artéria renal em crianças são hiperplasia fibromuscular e neurofibromatose. Hipocaliemia pode ocorrer em até 20% dos pacientes.os níveis de actividade da renina plasmática (ARP) encontram-se frequentemente no intervalo de referência, mas podem ser detectados níveis elevados de ARP após provocação com uma dose única de captopril 1 mg/kg., Pensa-se também que a isquemia Renal está subjacente ao hiperaldosteronismo secundário observado na hipertensão maligna.foram também notificados Hiperreninemia e aldosteronismo secundário em doentes com feocromocitoma, aparentemente como resultado de estenose funcional da artéria renal. Os tumores produtores de renina são muito raros e observam-se níveis muito elevados de ARP (até 50 ng/mL/h), frequentemente com um aumento da relação entre a pró-renina e a renina. Os tumores são geralmente de origem renal e incluem tumores de Wilms e carcinomas de células renais.,
hipercaliemia devido a insuficiência renal crónica também causa hiperaldosteronismo secundário. As baixas razões sódio-potássio podem ser medidas na saliva e nas fezes. A hipertensão induzida pela ciclosporina em doentes transplantados de órgãos sólidos pode também envolver um componente do hiperaldosteronismo.hiperaldosteronismo secundário na ausência de hipertensão ocorre como resultado de tentativas homeostáticas para manter a concentração de sódio ou o volume circulatório ou para reduzir a concentração de potássio., As condições clínicas em que ela pode surgir incluem diarréia, sudação excessiva, estados de baixa potência Cardíaca, e hipoalbuminemia devido a doença hepática ou renal ou síndrome nefrótica. O hiperaldosteronismo secundário também pode ocorrer de forma progressiva em recém-nascidos (ver abaixo).a dependência mineralocorticóide da reabsorção de sódio aumenta durante a infância e a infância, atingindo um pico no período neonatal antes de diminuir progressivamente com o avanço da idade., Este aumento ocorre porque a reabsorção de sódio e água pelo túbulo proximal é menos eficiente no início da vida, resultando num aumento da carga de sódio e água ao nível do túbulo renal distal.
Uma vez que a reabsorção de sódio e água do túbulo distal é mediada pelo eixo R-A-A, A ARP é aproximadamente 10 a 20 vezes mais elevada num recém-nascido do que num adulto., Consequentemente, os recém-nascidos apresentam aumentos relativos das taxas de produção de aldosterona (
300 µg/m2/dia vs 50 µg/m2/dia num adulto) e das concentrações plasmáticas de aldosterona (80 pg/dL vs 16 pg/dL). Estes aumentos no início da vida explicam por que razão os lactentes jovens apresentam sintomas clínicos profundos de hiperaldosteronismo que melhoram gradualmente com o avanço da idade.